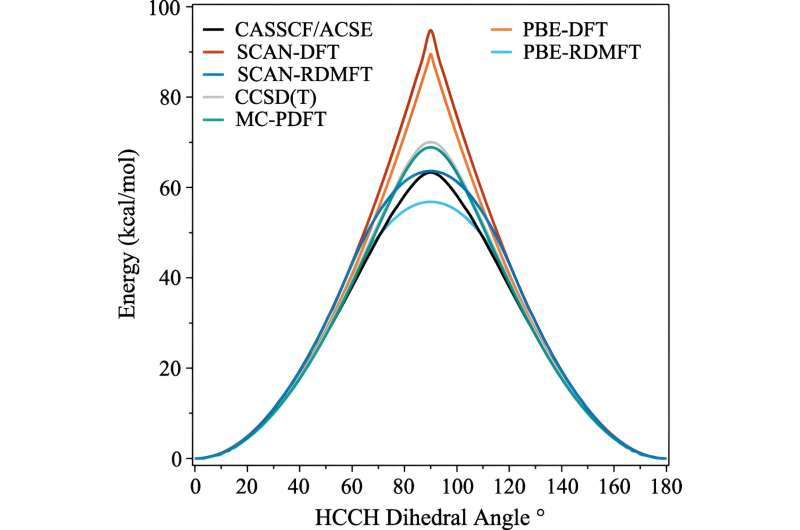
C2H4 Rotation barrier potential energy surface calculated from CASSCF(12,12)/ACSE, CCSD(T), PBE-RDMFT, SCAN-RDMFT, PBE-DFT, SCAN-DFT and CASSCF(12,12)/tPBE using cc – pVDZ basis set. Credit: Physical Review Letters (2023). DOI: 10.1103/PhysRevLett.131.243003
Like the humans who created them, computers find physics hard, but quantum mechanics even harder. But a new technique invented by three University of Chicago scientists could allow computers to simulate some of the challenging quantum mechanical effects in complex electronic materials with less effort.
By making these simulations more accurate and efficient, scientists hope the technique can help discover new molecules and materials, such as new solar cells or quantum computers.
“This advance has great potential to deepen our understanding of molecular phenomena, with major implications for chemistry, materials science and related fields,” said Daniel Gibney, a University of Chicago Ph.D. student in the Department of Chemistry. The first author of the paper, published on December 14 Physical Review Letters.
electrons and energy
A leaf or a solar panel looks smooth and simple from the outside, but zoom into the molecular level and you see an incredibly complex dance of electrons and molecules.
To achieve new advances in sustainability, manufacturing, agriculture, and many other fields, scientists model the behavior of these chemical and molecular interactions. This helps reveal the possibilities for a variety of new designs in the future, from new methods of carbon dioxide sequestration to new types of qubits.
Many advances have been made over the past few decades, but one area that remains difficult to simulate is where molecules are starting to exhibit complex quantum mechanical behavior that scientists call strong correlations.
The problem is that once electrons start showing off their most quantum mechanical effects, such as becoming “entangled,” calculations immediately require more computing power. Even supercomputers struggle to cope with these effects.
One of the commonly used calculations is called density functional theory. “This is basically the most common technique for predicting electronic structure, but it’s essentially an approximation where all electrons are treated as a function of one electron,” explains David Mazziotti, professor of chemistry and senior author of the study.
 give the relative energies of m- and p-phenylene with respect to o-phenylene. Image source: Physical Review Letters (2023). DOI: 10.1103/PhysRevLett.131.243003")
RDMFT and DFT using SCAN and PBE functionals, MC-PDFT using tPBE functional, and CCSD(T) give the relative energies of m- and p-phenylene with respect to o-phenylene. Credit: Physical Review Letters (2023). DOI: 10.1103/PhysRevLett.131.243003
For many calculations, approximations will do the job. But as electron behavior becomes more correlated, it starts to break down, just like what happens when quantum mechanics comes into play. In quantum mechanics, these electrons can be in multiple locations or orbits at the same time. This stymies not only the human brain, but also density functional theory.
“This is an important question because many of the issues we care about in the 21st century, such as new molecules and materials for renewable energy and sustainable development, require us to exploit the quantum properties of materials,” Mazziotti said.
Mazziotti, Gibney and third author Jan-Niklas Boyn discovered that they could add a general modification to density functional theory that entangles electrons in multiple orbits simultaneously.
“This allows the orbitals in the calculations to be not only completely filled or completely empty, but also somewhere in between,” Mazziotti said. “We get a single-electron picture that is still able to capture the relevant many-body electron effects. the resulting behavior.”
“Universal” adaptation
As a bonus, the scientists say, the code can be added to existing algorithms without having to rewrite the code. “Basically, fixes are initiated when needed but do not interfere with the rest of the code,” Gibney said.
It’s also versatile in that it can be added to codes that simulate the behavior of many kinds of electrons, whether it’s photovoltaic solar panels or carbon sequestration or superconducting materials or even biology.
For example, Boyne explained, one application might be in understanding the chemical reactions that occur using enzymes that contain metal atoms, called metalloenzymes.
“For example, there are a large number of metalloenzymes responsible for many chemical reactions in cells, but they are notoriously difficult to describe with current models,” he said. “This theory may in the near future allow us to solve this chemistry problem in a way that is currently impossible.”
More information:
Daniel Gibney et al., A general generalization of static correlation density functional theory, Physical Review Letters (2023). DOI: 10.1103/PhysRevLett.131.243003
Provided by University of Chicago
citation: New technology could make molecular modeling easier (2023, December 18), Retrieved December 24, 2023, from https://phys.org/news/2023-12-technique-molecules-easier. html
This document is protected by copyright. No part may be reproduced without written permission except in the interests of fair dealing for private study or research purposes. Content is for reference only.
#technology #molecular #modeling #easier
Image Source : phys.org