originate from B. Li et al. communication biology https://doi.org/10.1038/s42003-022-03353-5 (2022)
DNA methylation variation broadly reflects genetic variation, capturing population-level patterns that largely reflect global geography1,2,3,4; However, little is known about how genetic mixing shapes these relationships (but see Ref. 5,6). To address this gap, Li et al. analyzed DNA methylation variation in African Americans and found that by combining local genetic ancestry (LA) information, they could identify novel ancestor-specific genetic influences on DNA methylation. However, our reanalysis found that a significant proportion of their results involved methylation sites whose detection probe sequences contained at least one genetic variant that differed strongly between populations of European and African ancestry. Therefore, we hypothesize that many of the ancestor-specific signals reported by Li et al.is actually produced by differential hybridization efficiency7,8creating technological artifacts whose effects are confused with Los Angeles.
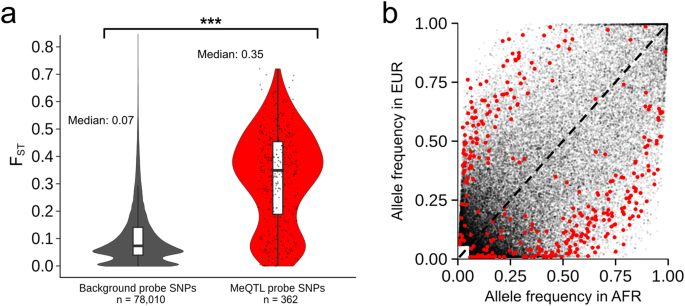
Li et al. 1284 cytosine-guanine dinucleotides (CpG) were identified, among which DNA methylation levels were correlated with LA.Note the high heritability of DNA methylation at these CpGs (average H2 45%), they then scanned methylation quantitative trait loci (meQTL), looking for single nucleotide polymorphisms (SNPs) associated with methylation levels within 1Mb of each LA-associated CpG. Under the traditional meQTL model, assuming the same effect size regardless of LA environment (i.e., LA-nave approach), they identified 1269 independent meQTL associations involving 946 unique CpGs. When they allowed meQTL effect sizes to vary between African and European haplotypes (i.e., LA-aware approach), they identified 1268 independent meQTL involving 135 unique CpGs, 152 of which showed significant effect size differences. Qualitative.However, by using a custom software package to carefully specify the detection area9 And cross-referencing with the comprehensive SNP set, we found some of the largest ancestor-specific effects identified by Li et al. Array probe sequences involving CpG sites contain common SNPs.Standard quality control procedures recommend filtering out such CpGs from analysis because probe sequence mismatches can affect hybridization efficiency and thus assay reliability.8. Furthermore, the probe SNPs we identified tended to differ strongly between populations of European and African ancestry (Fig. 1 ), a pattern we expected to lead to spurious LA-DNA methylation associations. Finally, we demonstrate that probe SNP-CpG site distance in base pairs strongly predicts meQTL effect size or LA-specific differences in effect size in the LA-nave and LA-aware methods, respectively (Fig. 2).This pattern would also be expected if the association was primarily driven by technical artifacts7. In light of our findings, we believe that Li et al.’s conclusion regarding the extensive LA-related heterogeneity in meQTL effect sizes should be re-evaluated.
A FYingshi Distributions between meQTL (red) and non-meQTL (grey) probe SNPs are significantly different (two-tailed t-test p-value=2.2e16, 95% confidence interval of the mean difference FYingshi [0.200.23]). Boxplots show the median (horizontal line), interquartile range (boxes), and 1.5 times the interquartile range (whiskers). Second The frequency of probe SNPs in AFR and EUR for the same probe SNP (red dots, meQTL from Li et al.; gray dots, non-meQTL) further emphasizes that they tend to differ significantly, i.e. they tend to deviate from the dashed line. The black line represents the same EUR and AFR allele frequency.

A MeQTL involving CpGs with probe SNPs (blue) have larger effect sizes than CpGs without probe SNPs (yellow) (95% confidence interval for the difference in mean effect size [0.28, 0.45]Two tails t-test p-value=1.44e15). Second Cubic regression of the distance between probe SNP and target CpG explained 42% of the variation in meQTL effect size for 351 CpG probes with SNPs (F-test p-Value for quadratic model = 0.0006). C LA-specific meQTL involving CpGs with probe SNPs (blue) have greater effect size differences (average) between European and African native ancestry than those without probe SNPs (yellow) 95% confidence interval for difference in effect size) [0.24, 0.70]Two tails t-test p-value=0.0002). d Quadratic regression of the distance between the probe SNP and the target CpG explained 25% of the variation in effect sizes for the 58 CpG probes with SNPs (F-test p-Value for linear model = 0.014).The boxplot is in A and C Shown are the median (horizontal line), interquartile range (boxes), and 1.5 interquartile range (whiskers).ribbon in Second and d Shows 2 standard errors above and below the model prediction.
We cross-referenced the probe sequences for all CpGs analyzed by Li et al. with a comprehensive set of variants likely to segregate in African American populations (see Methods section). We found that 37.5% of the meQTL-related CpGs in Li et al. contained at least one common probe SNP, while only 16.1% of CpGs not related to meQTL (non-meQTL CpGs). Then we calculated Cockerham and Weirs FYingshi Stage 3 between 1000 genomes10 African (AFR) and European (EUR) references for meQTL and non-meQTL probe SNPs (Fig. 1).We found that meQTL probe SNPs differed significantly more between African and European populations than non-meQTL probe SNPs (median FYingshi 0.35 vs. 0.07;Fig. 1). Therefore, if these meQTL-associated probe SNPs do affect hybridization efficiency, they would generate spurious associations between LA and DNA methylation.We found that for 10% (95/946) of the CpGs identified by the LA-nave method, affecting 18% of all LA-nave meQTL associations, the strongly divergent (i.e. minimal FYingshi 0.1; median FYingshi 0.39) Probe SNP directly affects the presence or absence of CpG sites. This represents a lower bound on the proportion of Li et al.’s results that are subject to technical artifacts, as individuals with CpG deletion variants would be incorrectly inferred to have low levels of DNA methylation.This proportion was even higher among CpGs identified in the meQTL group showing significant effect size heterogeneity; 22% (30/135) of CpGs affected 50% of these LA-specific meQTL associations (the lowest FYingshi 0.1; median FYingshi 0.44).
We then evaluated the relationship between meQTL effect size and CpG probe SNP distance.Previous studies have shown that the closer the probe SNP is to the target cytosine, the greater the impact on hybridization efficiency.7. First, as expected, we found that meQTLs with probe SNPs reported significantly higher effect sizes (and effect sizes) than meQTLs without probe SNPs (Fig. 2a, c). For the LA-nave results, we found that cubic regression best described the relationship between effect size and CpG-SNP distance (Fig. 2b). Importantly, under this model, CpG-SNP distance alone explained 42% of the variation in effect size. For the LA perception results, quadratic regression gave the best fit, with CpG-SNP distance explaining 25% of the variation in (Fig. 2d).This nonlinear relationship is driven by the much larger relative effect sizes of the SNPs at positions 0 and 18 Because these SNPs confuse loss of CpG genotype with loss of methylation.
As epigenetic research considers a greater range of human diversity, it will become increasingly important to assess the impact of genetic ancestry on DNA methylation. However, strict quality control measures must be taken to ensure that the technical characteristics of the array do not confound the analysis. We have shown that commercial DNA methylation array probe sequences contain genetic variants common in non-European populations, particularly African and African Americans. We believe that such CpGs should not be analyzed according to established quality control guidelines8. Rigorous probe exclusion is particularly important in the analysis of mixed populations, where multiple divergent genetic ancestors are combined in the study cohort. Our analysis identified LA-related CpGs identified by Li et al. Probe SNPs enriched for strong differences between European and African genetic ancestry. This may produce spurious correlations between DNA methylation levels and LA because these AFR and EUR haplotypes either do not bind the CpG probe sequences on the array with the same efficiency or they differ due to the presence of CpG . For at least 18% of the meQTL identified by the LA-nave method and 50% of the meQTL identified as LA-specific, the SNP directly affects the presence of CpG sites.However, as mentioned previously, SNPs throughout the probe sequence may bias effect estimates7,8. This bias is evident in our analysis, showing that the proximity of the probe SNP to the target cytosine alone explains 42% of the variation in the meQTL effect size estimated by Li et al.Although true biological effects tend to be stronger as SNP-CpG proximity increases, a recent large-scale analysis of 27 million samples cis-meQTL found that meQTLCpG distance explained <1% of the effect size variation on the order of 50 bp (i.e., the length of the probe sequence)11. Therefore, technological bias, rather than true biological effects, is the most likely driver of the patterns we observed.
In conclusion, we caution against correcting local ancestry in future meQTL studies unless technical biases are strictly considered. This must include cross-referencing methylation probe coordinates with a full set of common variants, such as the 1000 Genomes panel10. To this end, we propose probeSNPffer9, a tool that performs this cross-reference step (see Methods section). We hypothesize that many variants in the reference SNP list were excluded from consideration by Li et al.’s stringent quality control criteria but are still likely to be present in their sample. For example, in 1000 genome ASW, more than 14 million variants segregate at >1% frequency (n=61), while Li et al. Only 4.7 million variants with frequency >1% were retained in the larger sample (n=1031). A large proportion of these nearly 10 million deletion variants will certainly segregate within samples, and this must be considered when filtering CpGs, especially when seeking to understand the impact of local ancestry on DNA methylation in mixed populations.
#Genetic #differentiation #probe #SNPs #leads #spurious #results #meQTL #discovery #Communications #Biology
Image Source : www.nature.com